IPLab:Lab 5:Gaucher Disease
Contents
Clinical Summary[edit]
This 23-year-old female went to her doctor because of chronic fatigue, bone pain, and easy bruising and frequent nose bleeds. Physical exam demonstrated hepatomegaly and splenomegaly and lab results demonstrated anemia and thrombocytopenia. Biopsy and further workup led to a diagnosis of Gaucher disease.
Despite appropriate therapy the anemia and thrombocytopenia persisted as well as the severe splenomegaly. A splenectomy was performed. The surface of the spleen was pale and roughly granular. The cut surface revealed the same pale appearance.
Images[edit]

This is a gross photograph of spleen from this case. The spleen is enlarged and the surface is finely granular.

This is a cut section of spleen from this case. Again note the fine granular appearance to the tissue.
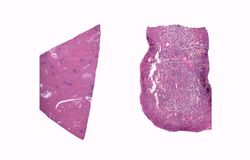
This is a low-power photomicrograph of normal spleen (left) and the spleen from this case (right). The loose appearance of the tissue in the Gaucher spleen is due to artifactual loss of tissue during histologic processing.
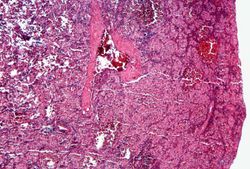
This is a photomicrograph of the spleen from this case. There is very little if any white pulp evident in this section.
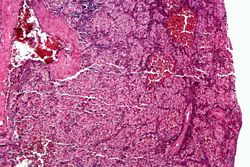
This is a higher-power photomicrograph of the spleen from this case. Again there is no white pulp and the red pulp is filled with large eosinophilic cells.
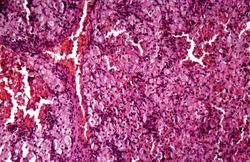
This is another high-power photomicrograph of the spleen from this case. At this power it is easier to see the large eosinophilic cells.
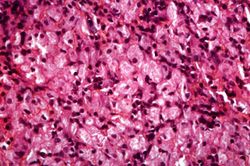
This is another high-power photomicrograph of the spleen from this case. At this high power individual cells can be better appreciated.
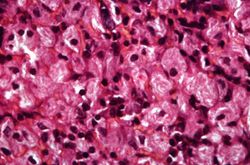
This is a higher-power photomicrograph of the spleen from this case. At this higher power individual cells can be better appreciated and the fibrillar nature of the eosinophilic cytoplasmic material can be seen.








Virtual Microscopy[edit]
Study Questions[edit]
Additional Resources[edit]
Reference[edit]
- eMedicine Medical Library: Gaucher Disease
- Merck Manual: Splenomegaly
- Merck Manual: Lysosomal Storage Disorders
Journal Articles[edit]
- Barone R, Pavone V, Nigro F, Chabàs A, Fiumara A. Extraordinary bone involvement in a gaucher disease type I patient. Br J Haematol 2000 Mar;108(4):838-41.
Images[edit]
| |||||
Trauma (from fracture or surgery) is one cause of asceptic (or avascular) necrosis of bone, which is defined as the death of bone and bone marrow in the abscence of an infectious agent.